Our Research
Multiscale Computational Chemistry
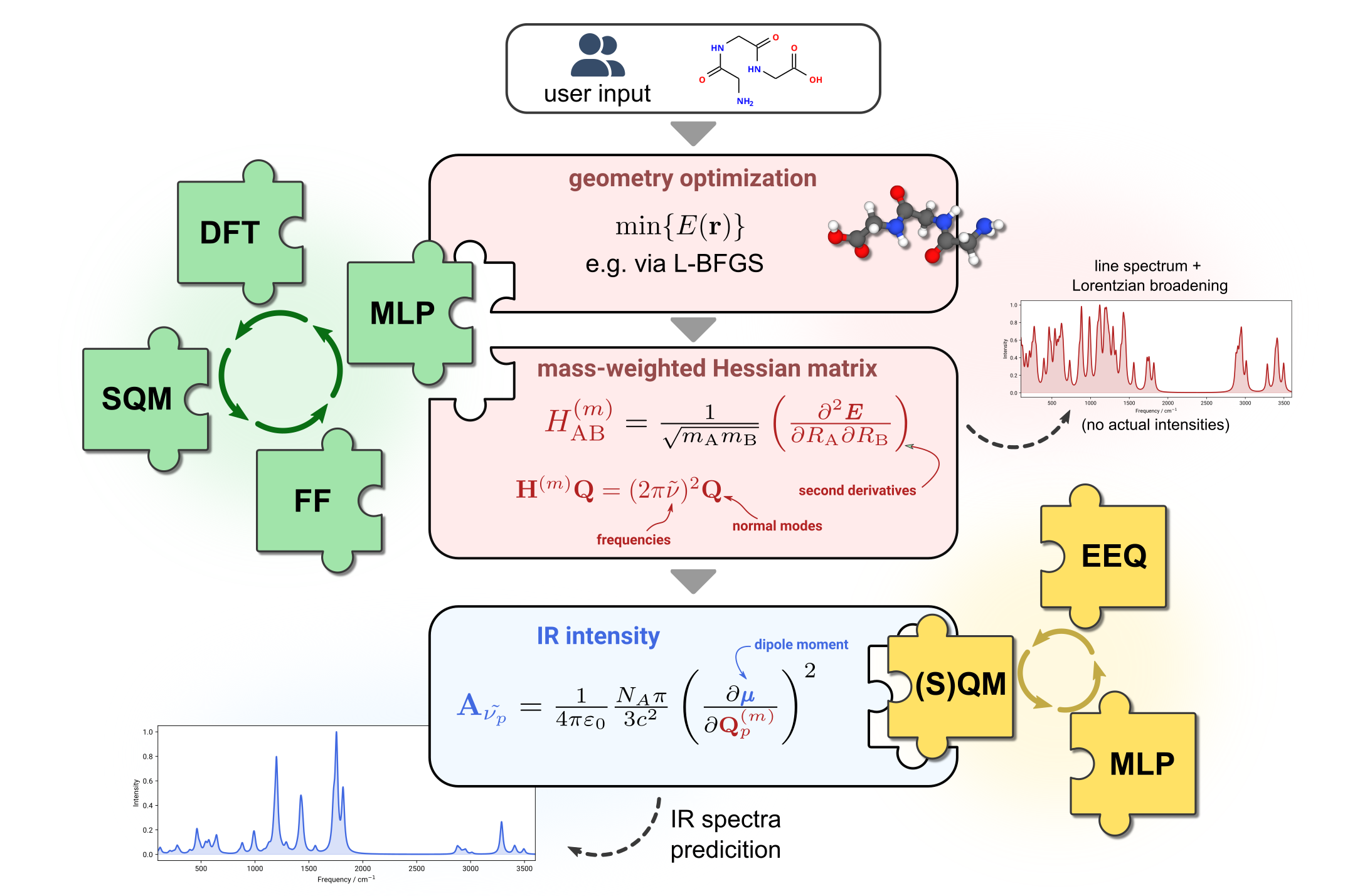
Our research explores the application of so-called multiscale or multilevel protocols in computational chemistry, which combine methods tailored to describe different regimes of chemistry within a single hierarchical framework. By combining semiempirical quantum mechanics for low-cost efficient electronic structure calculations, classical force-fields for sampling applications, and modern machine learning potentials for rapid and adaptable approximations, these protocols enable us to efficiently study complex molecular systems across different scales of accuracy and computational cost.
A key focus in our group is on molecular thermodynamics, tied to the investigation of the molecular free energy landscape. The free energy landscape provides insights into the stability and dynamics of molecular systems across all chemical disciplines. In the context of computational spectroscopy, we employ multiscale methods to study vibrational (IR, Raman) and NMR spectra, targeting the interpretation of experimental data and the identification of molecular systems.
Multiscale protocols developed in our group make computational techniques accessible to large molecular systems and enable high-throughput studies, which benefits progress in both chemical discovery and computational analytics.
Chemical Space Exploration
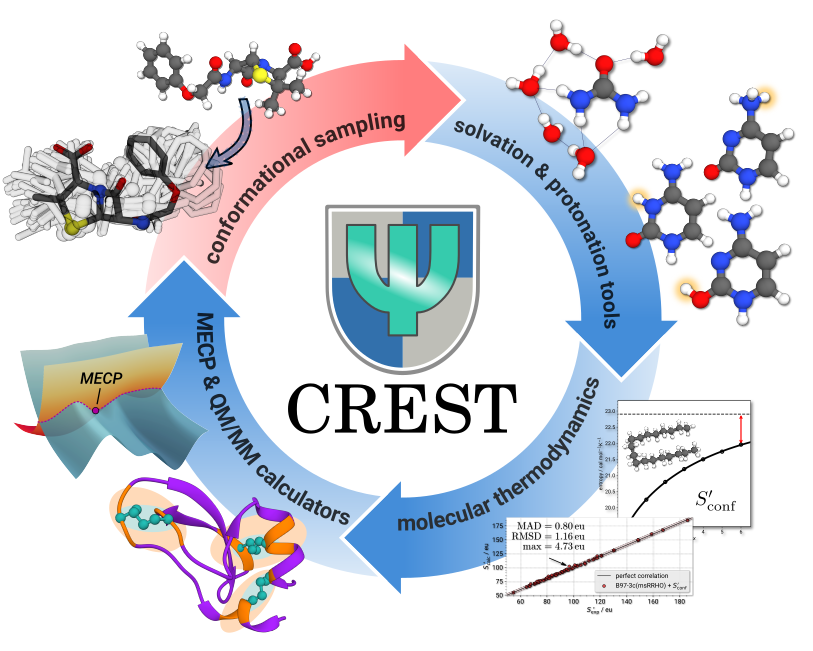
Our research on exploration algorithms focuses on development of advanced sampling techniques to investigate the low-energy chemical space of molecular systems. By leveraging the aforementioned hierarchical workflows, we address challenges of efficiently exploring the vast energy landscapes of chemical systems, uncovering the multitude of structural effects like various kinds of isomerism and associated reactions that define the molecular behavior.
A particular emphasis lies on automation and accessibility, enabling researchers to streamline workflows and explore chemical space and chemical reactivity with minimal manual intervention. This is especially critical for conformational sampling, a foundation for almost all computational studies involving non-rigid molecular systems. Accurate and efficient exploration of molecular conformations is essential for predicting thermodynamic properties, chemical reactivity, and intermolecular interactions. To support these goals, we develop open-source tools such as the popular CREST program (https://github.com/crest-lab/crest).
Computation-Aided Chemical Analytics

A key interest in our group is to explore how computational methods can help identify and analyze chemical compounds, mixtures, or impurities from experimental measurements. Here, we employ the above mentioned automated workflows combining multiscale modeling, quantum mechanical simulations, and machine learning procedures to enhance chemical analysis.
By integrating techniques like IR, NMR, and mass spectroscopy with computational predictions, we aim to provide scalable, cost-effective solutions for identifying complex samples. Our research addresses key questions about the limits and capabilities of computational approaches, supporting conventional analytic chemistry with robust insights and methodologies from theory and thus bridging experimental and computational research.
Software


